2026 |

Anika Neureiter, Uniklinik RWTH Aachen
Conference: ISSCR 2026 Annual Meeting
Abstract Title: Patient-Specific iPSC-Derived Sensory Neurons as a Gateway to Individualized Neuropathic Pain Modelling and Treatment
2025 |

Marina Hommersom, Radboud University Medical Center
Conference: AXON2025
Abstract Title: Human neuronal networks on micro-electrode arrays as a tool to assess genotype-phenotype correlation in CACNA1A-related disorders

Claire Hetherington, The University of Liverpool
Conference: Encephalitis 2025
Abstract Title: HSV-1 infection drives neuronal network hyperexcitability and hypersynchrony in an in vitro model of seizures in herpes simplex encephalitis

Feline Benavides, Erasmus University Medical Center
Conference: The European Scientific Working Group on Influenza (ESWI)
Abstract Title: Cell Tropism, Replication Dynamics, and effect on neural network activity to Seasonal and Pandemic Influenza A Virus Infection in a hiPSC-Derived Neural Co-Culture Model

Kenneth Wilson, Buck Institute for Research on Aging
Conference: Cold Spring Harbor Laboratory Symposium: Senescence & Aging
Abstract Title: OXR1 regulates cellular senescence and neuronal aging through retromer-mediated actin branching

Laura Mosqueira, IIS Biogipuzkoa
Conference: Rome MEA User Meeting
Abstract Title: Bioelectric platforms for skeletal muscle and cardiac in vitro models: phenotypic characterization and evaluation of MP-001 activity

Yao Yin, Shanghaitech University
Conference: Cell Symposium: Engineering development and disease in organoids
Abstract Title: Generation of human PSCs-derived multi-tissue organoids

Adam Pavlinek, King's College London
Conference: Organization for the Study of Sex Differences (OSSD) Annual Meeting
Abstract Title: Using organoids to study neurodevelopmental sex differences

Caleb Aguayo, Wake Forest Institute for Regenerative Medicine
Conference: American Society for Pharmacology and Experimental Therapeutics (ASPET)
Abstract Title: Human Neuromuscular Junction Platform for Investigating Pathogen and Toxin-Induced Dysfunction in Disease and Drug Discovery

Alia Arslanova, Simon Fraser University
Conference: Heart Rhythm 2025
Abstract Title: Exploring the molecular underpinnings of a large genomic deletion in cardiac RYR2 gene
2024 |

Juran Choe, Daegu Gyeongbuk Institute of Science and Technology
Conference: 2024 KSBNS
Abstract Title: Investigating the excitatory-inhibitory imbalance in Alzheimer's disease using fusion organoids

Bianca de Freitas Brenha, Case Western Reserve University
Conference: AES 2024 Annual Meeting
Abstract Title: A Multifaceted High-Content Screening platform for Analgesic Compounds Using Human Induced Pluripotent Stem Cell-Derived Nociceptors

Saif Dababneh, University of British Columbia
Conference: 2024 Till & McCulloch Meetings
Abstract Title: Inducible arrhythmias in a human iPSC model for calcium release deficiency syndrome
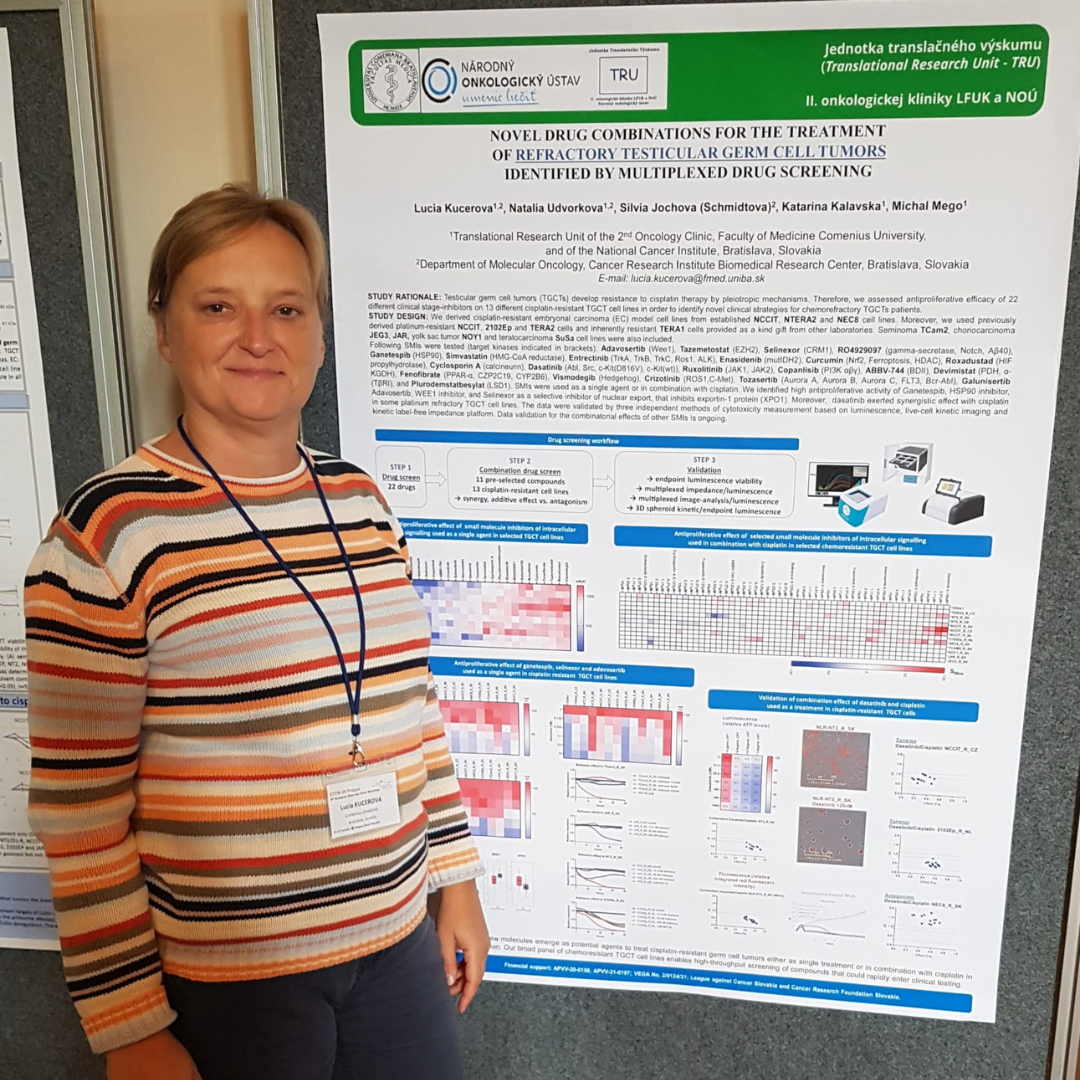
Lucia Kucerova, Comenius University
Conference: 10th European Testicular Tumor Workshop
Abstract Title: Novel drug combinations for the treatment of refractory testicular germ cell tumors identified by multiplexed drug screening
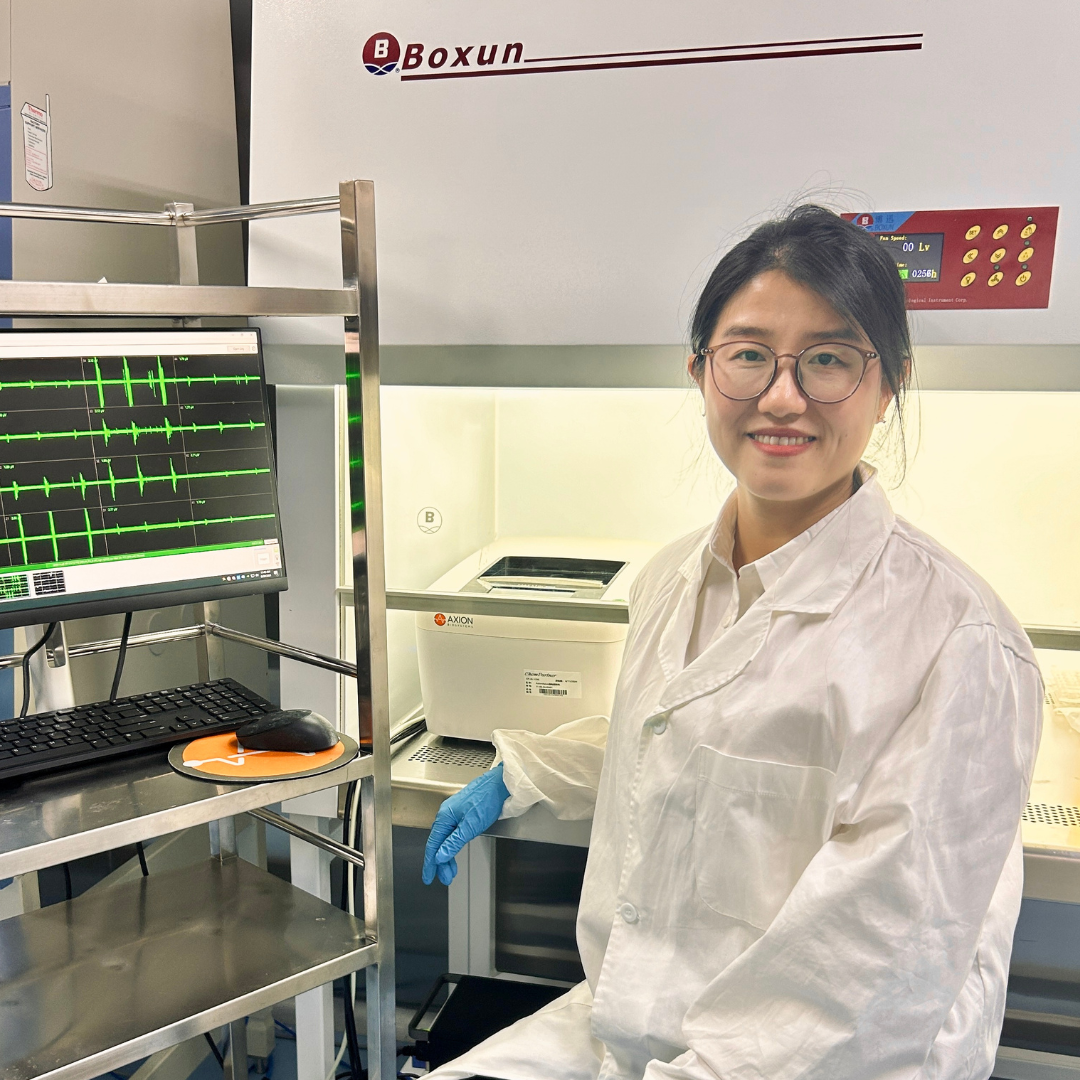
Lulan Chen, ChemPartner
Conference: Chinese Neuroscience Society 2024
Abstract Title: A higher efficient in vitro functional assay for anti-epileptic drug discovery

Pang Wei, ShanghaiTech University
Conference: Cell Symposia: Engineering development and disease in organoids
Abstract Title: Generation of human region-specific brain organoids with medullary spinal trigeminal nuclei

Wanhao Chi, Northwestern University
Conference: Society for Neuroscience (SfN) 2024
Abstract Title: Gain of function of KCNH1 induces hypoexcitability in cortical excitatory neurons derived from human induced pluripotent stem cells

Elliot Mock, University of Oxford
Conference: EMBO Workshop Autophagy across scales
Abstract Title: Pharmacological correction of excessive basal mitophagy by pathological levels of α-synuclein restores neuronal function in human dopaminergic neurons

Kennedy Barkhouse, The Hospital for Sick Children
Conference: Canadian Association for Neuroscience 2024
Abstract Title: Interrogating the effects of human herpesvirus 6 infection on neurons and glia derived from human pluripotent stem cells

Xuemei Huang, Wuhan University
Conference: 海南MPS, Hainan
Abstract Title: Trans-organoid co-differentiation (TOCO) chip for constructing heart-brain developmental model

Giada Cattelan, Eurac Research
Conference: 13th Congress of The International Society for Autonomic Neuroscience (ISAN)
Abstract Title: In vitro modelling of the neurocardiac junction with a novel human iPSC-based co-culture system: new perspectives for the investigation of cardiac autonomic regulation

Marie Le Bihan, RadboudUMC
Conference: Federation of European Neuroscience Societies (FENS) Forum 2024
Abstract Title: Unraveling mTORopathies: mTOR hyperactivation induces mutation-specific functional phenotypes in human neuronal networks

Sofia Puvogel, RadboudUMC
Conference: Federation of European Neuroscience Societies (FENS) Forum 2024

Marine Tessier, Aix Marseille Université
Conference: Federation of European Neuroscience Societies (FENS) Forum 2024

Kyndra Higgins, University of Georgia
Conference: Gordon Research Conference: Non-Cell-Autonomous Mechanisms in Normal Mammary Gland and Breast Cancer Development
Abstract Title: Utilizing In Vitro Bioelectronic Assays in Cancer Cell Characterization

Curran Landry, Case Western Reserve University
Conference: Keystone Symposium: Neurodegenerative Diseases
Abstract Title: VAPB P56S Variant Sensitizes ALS Patient-Derived Motor Neurons to the Integrated Stress Response

Adham Farah, University of Oxford
Conference: OBN BioTrinity 2024
Abstract Title: Using Micro-electrode Array (MEA) to test the impact of patient autoantibodies on sensory neuron excitability

Hsueh-Fu Wu, University of Georgia
Conference: 2024 Spring Brain Conference
Abstract Title: A modular platform to generate functional sympathetic neuron-innervated heart assembloids

Caitlyn Gaffney, MD Anderson Cancer Center
Conference: International Association for the Study of Pain
Abstract Title: Inflammatory Neuropathy in Mouse and Primate Models of Colorectal Cancer

Ilka Scharkin, Leibniz Research Institute for Environmental Medicine
Conference: 5th International Conference on Developmental Neurotoxicity Testing (DNT5)
Abstract Title: Refinement of stem cell-based in vitro assays towards a regulatory use for developmental and adult neurotoxicity testing of chemicals

Vipendra Kumar, University of Illinois Urbana-Champaign
Conference: Gordon Research Conference on Fragile X and Autism-Related Disorders, Advances in Understanding Disease Mechanisms and Translation to Therapeutics
Abstract Title: Role of mGluR7 in Fragile X syndrome

Madeleine Strom, Purdue University
Conference: Society of Toxicology Annual Meeting 2024
Abstract Title: Developmental Exposure of Human Induced Pluripotent Stem Cell-derived Cortical Cultures to Methylmercury Induces Persistent Functional Effects.

Sarah Jolly, University College London
Conference: 6th Neuroimmunology Drug Development Summit
Abstract Title: Astrocytes as drug targets in Alzheimer's disease.
2023 |

Fiona Ducotterd, University College London
Conference: Society for Neuroscience 2023
Abstract Title: Validation of astrocytic cAMP signaling to study therapeutic targets for Alzheimer's disease

Laura-Sophie Frommelt, Eurac Research
Conference: CBFH Biennial Meeting 2023
Abstract Title: Green chemistry, red flags: multiparametric cardiotoxicity screening of phytochemicals using hiPSC-CMs-MEA Assay

Chia-Wei Huang, The University of Georgia
Conference: The 2023 Meeting of The Society for Glycobiology
Abstract Title: Dysregulated O-GlcNAcylation is a molecular link to Alzheimer's Disease

Kristina Bartmann, IUF - Research Institute for Environmental Medicine
Conference: 12th World Congress on Alternatives and Animal Use in the Life Sciences
Abstract Title: Assessment of human neural network formation and function using 2D and 3D hiPSC-derived cell systems

Navroop Kaur Dhaliwal, SickKids
Conference: International Society for Stem Cell Research (ISSCR) 2023
Abstract Title: Investigating the mechanism of PTEN mutation-related neurodevelopmental disorders in human pluripotent stem cells-derived 2D and 3D neural cultures

James Burford, University of Southern California
Conference: International Society for Stem Cell Research (ISSCR) 2023
Abstract Title: Functional characterization of a human pluripotent stem cell-derived cerebellar organoid model of RNA exosome-linked pontocerebellar hypoplasia type 1B

Lora-Sophie Gerber, Utrecht University
Conference: Meeting of the International Neurotoxicology Association (INA) 2023
Abstract Title: Neurotoxicity of non-volatile and semi-volatile diesel exhaust-derived ultrafine particles

Heather Lin, Emory University
Conference: ASGCT 2023
Abstract Title: Engineering Optimal CAR T Cells to Overcome Pancreatic Tumors with Secreted Antagonistic Peptides

Lucy Granat, University College London
Conference: Neurodegeneration: New biology guiding the next generation of therapeutic development
Abstract Title: Development of an astrocyte platform to support the validation of astrocytic cAMP signaling as a therapeutic target for AD

Zeinab Hamze, PhD, INSERM/Aix-Marseille University
Conference: Peripheral Nerve Society 2023
Abstract Title: Pathophysiological mechanisms in a new form of Charcot Marie Tooth due to a mutation in PDXK

Hadar Bootz-Maoz, Bar Ilan University
Conference: Cell Symposia: Infection biology in the age of the microbiome
Abstract Title: Diet-induced modifications to human microbiome reshape colonic homeostasis in irritable bowel syndrome

Anke Tukker, Purdue University
Conference: 62nd Annual meeting of the Society of Toxicology
Abstract Title: Early-life MeHg exposure of human iPSC-derived cortical cultures persistently alters the homeostatic
neuronal state

Emma Kasteel, Utrecht University
Conference: 62nd Annual meeting of the Society of Toxicology
Abstract Title: In vitro neurotoxicity of micro- and nanoplastics measured using microelectrode array (MEA) recordings in rat cortical cultures
Devon Guerrelli, Children's National Hospital
Conference: 2023 Gordon Research Conference: Cardiac Arrhythmia Mechanisms
Abstract Title: Off-Label Drugs in Cardiology: Developing a Pediatric Preclinical Model to Assess Arrhythmogenesis

Sarah Jolly, ARUK-UCL Drug Discovery Institute; UCL
Conference: ADPD 2023, Gothenburg
Abstract Title: Modelling of glia-neuron crosstalk in vitro to facilitate drug discovery for Alzheimer’s disease
2022 |

Jacob Adelman, Medical College of Wisconsin
Conference: Neuroscience 2022, San Diego
Abstract Title: Evaluating the influence of HCMV infection on Alzheimer's Disease pathology

Hamed Salmanzadeh, University of the Pacific
Conference: New York Stem Cell Foundation Conference 2022
Abstract Title: Properties of Human Stem Cell-derived Neurons in Long-term Cell Culture as a Model for Antiseizure Drug Discovery

Zhefu Que, Purdue University
Conference: Neuroscience 2022, San Diego
Abstract Title: Regulation of hyperexcitable neuron carrying epilepsy-associated sodium channel Nav1.2-L1342P variant by hiPSC-derived microglia

Maria Isabel Olivero Acosta, Purdue University
Conference: Neuroscience 2022, San Diego
Abstract Title: Modeling epilepsy-related SCN2A gain-of-function mutation L1342P with CRISPR-edited human-induced

Emma Dyke, RadboudUMC
Conference: EMBO Cilia2022
Abstract Title: Investigating the role of ciliary proteins in hiPSC-derived excitatory neurons

Emily Welby, Medical College of Wisconsin
Conference: Cure SMA 2022 Research and Clinical Care Meeting
Abstract Title: The Role of Astrocytes in Spinal Muscular Atrophy Motor Neuron Synaptic Defects

Marina Hommersom, Radboudumc
Conference: FENS 2022, Paris Expo Porte de Versailles
Abstract Title: Electrophysiological characteristics of human induced pluripotent stem cell-derived neurons with CACNA1A variants

Hamed Salmanzadeh, University of the Pacific
Conference: ISSCR, San Francisco
Abstract Title: Properties of Human Stem Cell-Derived Neurons in 2D and 3D in Long-term Cell Culture

Pascal Röderer, University Hospital Bonn
Conference: IASP 2022 World Congress on Pain, Canada
Abstract Title: Forward programmed human sensory neurons for modeling genetic pain disorders

Shannon McDonnell, Queen's University Belfast
Conference: Young International Cell Senescence Association Conference, United Kingdom
Abstract Title: Divergent autocrine roles for the endothelial SASP
Kartik Pradeepan, Robarts Research Institute/Western University
Conference: Gordon Research Seminar/Conference for Fragile X and Autism Related Conditions, Italy

Bettina Weigel, German Cancer Research Center
Conference: Neural stem cells: From basic understanding to translational applications, Greece
Abstract Title: MYT1L haploinsufficiency causes treatable mental disease phenotypes in mice and human neurons
Katy Hole, University of Bath
Conference: ADPD 2022 Advances in Science and Therapy in Barcelona, Spain
Abstract Title: Overexpression of an FTLD mutant of Tau disrupts the Tau Interactome and enhances network excitability in primary cortical neurons
2021 |

Ujwal Boddeti, University of Maryland
Conference: Congress of Neurological Surgeons Annual Meeting, Austin, Texas
Abstract Title: Role of functional network formation in a stimulation model of cortical epilepsy
2020 |

Muhamad Mergaye, Ohio State University
Conference: 14th Annual Conference on Stem Cell and Biomaterials, Paris, France
Abstract Title: Functional assessment of low oxygen preconditioned human iPSC-derived cardiomyocytes subjected to anoxia challenge
Kristina Bartmann, IUF-Leibniz Research Institute
Conference: 2020 Society of Toxicology Annual Meeting, Anaheim, CA
Abstract Title: A human iPSC-based in vitro neuronal network formation assay for regulatory developmental neurotoxicity testing

Sylmarie Davila-Montero, Michigan State University
Conference: 2020 Society of Toxicology Annual Meeting, Anaheim, CA
Abstract Title: Improving identification of neuroactive compounds using temporal information from microelectrode array recordings of cortical neural networks and a semi-supervised classification algorithm

Anne Zwartsen, Utrecht University
Conference: International Neurotoxicology Association 17 Meeting, Duesseldorf, Germany
Abstract Title: Is the decrease in in vitro neuronal activity reversible following acute and prolonged exposure to new psychoactive substances (NPS)?
2019 |
Devon Guerrelli, Children's National Medical Center
Conference: 2019 BMES Annual Meeting, Philadelphia, PA
Abstract Title: Assessing the toxicity of medical device plasticizers on cardiac gap junction intracellular communication

Maria Jose Quezada, Northwestern University
Conference: Society for Neuroscience 2019, Chicago, IL
Abstract Title: Phenotypic characterization of human iPSC-derived motor neurons that carry the common val66met single nucleotide polymorphism of brain derived neurotrophic factor gene using multielectrode array and an in vitro stretch injury system
Kartik Pradeepan, Western University, Canada
Conference: Society for Neuroscience 2019, Chicago, IL
Abstract Title: Developmental population-level differences of stem cell derived excitatory networks from SHANK2 ASD patients

Morgan Zipperly, University of Alabama at Birmingham
Conference: Society for Neuroscience 2019, Chicago, IL
Abstract Title: The role of Gadd45b in striatal physiology and cocaine reward

Lydia Sibley, Shirley Ryan Ability Lab
Conference: Society for Neuroscience 2019, Chicago, IL
Abstract Title: Establishing a co-culture model of human induced pluripotent stem cell-derived motor neurons and primary stem cell-derived myotubes from patients with cerebral palsy

Derek Tai, Center for Genomic Medicine, Massachusetts General Hospital
Conference: Society for Neuroscience 2019, Chicago, IL
Abstract Title: Deciphering the molecular basis of neuronal development deficits in the recurrent genomic disorder

Danielle Tomasello, Whitehead Institute for Biomedical Research
Conference: Human Genome Meeting, Seoul, South Korea
Abstract Title: FAM57B Ceramide Synthase, a Hub Gene within 16p11.2 Deletion Syndrome, Maintains Physiological Lipid Membrane Composition
Katherine Savell, University of Alabama at Birmingham
Conference: American Society of Gene & Cell Therapy, Washington, DC
Abstract Title: CRISPR-Based Transcriptional Control of Dopaminergic Gene Programs

Xiaofeng Ma, Washington University School of Medicine
Conference: 66th Annual Meeting of the Society for Reproductive Investigation, Paris, France
Abstract Title: Use of multi-electrode array recordings in studies of electric activities of the uterine smooth muscle of mice
2018 |

Rebecca Mok, The Hospital for Sick Children & University of Toronto
Conference: Society for Neuroscience 2018, San Diego, CA
Abstract Title: Altered connectivity in Rett syndrome stem cell-derived neural networks

Bryan James Black, The University of Texas at Dallas
Conference: Society for Neuroscience 2018, San Diego, CA
Abstract Title: Human induced pluripotent stem cell (hiPSC)-derived motor neurons co-cultured with primary astrocytes exhibit functional network sensitivity to botulinum neurotoxin

Michael Nestor, The Hussman Institute for Autism
Conference: Society for Neuroscience 2018, San Diego, CA
Abstract Title: High throughput screening of hiPSC derived brain organoids to probe for phenotypes in idiopathic autism

Dina Simkin, Northwestern University
Conference: Society for Neuroscience 2018, San Diego, CA
Abstract Title: Dyshomeostatic mechanisms of KCNQ2-related epileptic encephalopathy in patient-specific iPSC-derived excitatory neurons

Ashwini Sri Hari, University of Colorado
Conference: Society of Toxicology 2018, San Antonio, TX
Abstract Title: Thiol-containing compounds attenuate oxidative stress and neuronal hyperexcitability in vitro