Author: Jim Ross, Ph.D.
Lab+Life Scientist, 2021
Amyotrophic lateral sclerosis (ALS) is a disease of the brain affecting approximately one in every 50,000 people. Symptoms tend to set in as people reach middle age, causing a decline in motor control and the progressive loss of ability to speak, eat and breathe, leading to death.
No part of ALS is subtle, yet no diagnostic criteria have been established, and the drugs available for treatment only treat symptoms and slow patients’ decline. But ALS research is making strides towards uncovering treatments to reduce the damage ALS inflicts on people’s lives. In particular, the work carried out in the lab of Evangelos Kiskinis, PhD, Assistant Professor of Neurology at Northwestern University Feinberg School of Medicine, seeks to create better models of neurological diseases, like ALS, with induced pluripotent stem cells (iPSCs). His work, and that of his team while still at Harvard, created the first iPSC model of ALS which was used to screen potential treatments and led directly to a phase 2 clinical trial to investigate a candidate drug’s potential as a desperately needed treatment for ALS.
Limitations of classic electrophysiology techniques
As is the case when addressing any neurological disease, scientists studying ALS must overcome a significant barrier: the human brain is a complex organ, a veritable cacophony of intercommunicating cells, difficult to recreate with animal models and even more difficult to measure consistently in a lab setting. Motor neurons, derived by Kiskinis and his team, from iPSCs from ALS patients were shown to recapitulate many of the molecular pathways of the disease. By combining faster, easier electrophysiology methods like bioelectronic assays with traditional methods like patch-clamp, researchers can validate the mechanistical underpinnings of an electrophysiological disease phenotype and screen against it. In the end, these results can help them decipher the disease and identify treatments to reverse or even cure it.
Manual patch-clamp is a powerful but difficult technique used to illuminate the mechanisms of neurological disease by measuring how individual neurons or ion channels behave electrically. In a typical whole-cell patch-clamp electrophysiology experiment in vitro, a neuroscientist finds a single neuron in a dish, seals a micropipette to the membrane to rupture it, and measures voltage or current in response to stimuli. Time is of the essence: because the cell membrane is ruptured by the pipette, readings can only be taken for several minutes before the cell dies. Moreover, each experiment is time-consuming: even an experienced electrophysiologist may only be able to measure a handful of neurons per day.
Ultimately, patch-clamp methods are designed to examine one cell at a time. To illuminate how variations in genetics, environment and drug exposure affect the behaviour of neurons, however, it’s important to look at electrical activity across a network of cells.
Getting a broader perspective with microelectrode arrays
Measuring from single cells is informative in the study of ALS, but it is not enough. To test the many genetic mutations associated with the disease and screen which therapies may be effective, scientists need new and faster ways to functionally test their neuronal models. Researchers are finding that this information can be gained through microelectrode array (MEA) technology.
A typical MEA experiment is run by culturing cells in a multiwell plate that has tightly packed microelectrodes embedded in the bottom of each well. In the wells, neurons form networks (Figure 1). A single experiment can simultaneously measure changes in the electrical activity of the network of cells over time and across multiple replicates or multiple variables. Unlike classic electrophysiology methods, the technique is remarkably simple to execute. Anyone trained in basic cell culture techniques can run an MEA assay and access a wealth of electrophysiological information to model brain activity and provide context to better understand data gained through classic single-cell techniques.
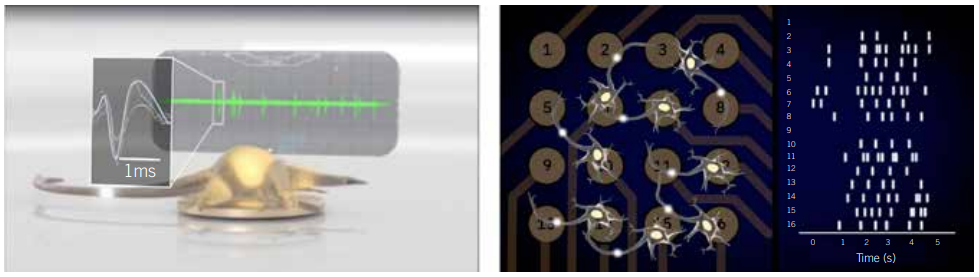
Figure 1: Example voltage trace of an individual neuron (left) or a raster plot of networks activity (right) in an MEA plate. Sensitive electrodes record the voltage 12,500 times/s, capturing neuronal firing in real time (left). By comparing the firing across the electrodes, network patterns emerge.
A combination approach to study familial ALS
The causes underlying ALS are complex, but a subset of cases is familial: associated with mutated SOD1 genes. To understand how these mutations of SOD1 influence phenotype, Kiskinis and his team generated patient-specific ALS-in-a-dish models, by culturing motor neurons derived from the iPSCs of ALS patients carrying the SOD1 A4V mutation (SOD1A4V). Kiskinis used a combination of MEA and manual patch-clamp electrophysiological techniques to characterise these models. They showed that hyperexcitability observed in clinical neurophysiological studies is recapitulated in iPSC-derived motor neurons, as measured by increased activity in MEA and confirmed with patch clamp. Due to the higher throughput nature of MEA, ultimately, they were able to show that a drug, ezogabine (retigabine), had the potential to reverse that hyperexcitability in cultured cells and in people.
In a multiwell MEA assay, the scientists were able to simultaneously gather several key pieces of information. They confirmed that the SOD1A4V motor neurons were hyperexcitable compared to control cells. Blocking inhibitor neurons in the control condition did not increase their excitability, suggesting that the SOD1A4V cells’ phenotype wasn’t due to a defect in inhibitory neurons. Finally, they observed that correcting the A4V mutation of SOD1A4V motor neurons could correct the hyperexcitability.
Actionable results
The SOD1 A4V mutation affects oxidative stress in motor neurons. Kiskinis and his team discovered a link between this stress in the endoplasmic reticulum and ion channel function in neurons. To gain a deeper understanding of how the mutation affects signalling, the Kiskinis lab ran patch-clamp assays and discovered SOD1A4V motor neurons have reduced delayed-rectifier potassium currents. Through a combination of patch-clamp and MEA assays, the team demonstrated that the cells’ hyperexcitability could be reduced and their survival times improved by treating them with a drug that reverts the activity of the potassium channels to normal.
These in vitro results offered a potential avenue to finally treat patients. By screening approved compounds for off-label use, they were cleared to jump straight to a phase 2 clinical trial, which showed that ALS patients treated with ezogabine over the course of two months exhibited a reduction in neuronal hyperexcitability. This phase 2 study laid important foundations for future clinical work, establishing that neuronal hyperexcitability may be a pharmacodynamic biomarker. Future clinical trials based on these findings still need to determine if reduced hyperexcitability is truly correlated with prolonged lifespans and reduction of symptoms.
Kiskinis’s studies demonstrate how MEA technology can put neuroscience research on the fast track towards clinically meaningful results. Moreover, because the technology does not call for extensive training to use, more labs within the scientific community have the potential to leverage it in their research. In all, by using a combination of single-cell techniques and MEA approaches that provide scalability and network-level insight, scientists can attain a sufficiently complex understanding of the disease, identify biomarkers, and gain traction in the search for treatments and cures that have eluded medical science so far.
*Dr Jim Ross is the Chief Technology Officer and co-founder of Axion Biosystems, a company that develops instrumentation to support bioelectronic assays in the life sciences. Bioelectronic assays allow scientists to precisely and continuously monitor cultured cells without labels or dyes


