Introduction
Amyotrophic lateral sclerosis (ALS), Lou Gehrig’s disease, is a fatal neurodegenerative disease of the motor nervous system. Motor neurons are located in the brain, brain stem, and spinal cord and control communication between the central nervous system and voluntary muscles of the body. Although many distinct mutations in a variety of genes are known to cause ALS, it remains poorly understood how they impact motor neuron biology and whether they converge on common pathways to cause neural degeneration. Researchers at Harvard University utilized induced pluripotent stem cells (iPSCs) to generate neurons from ALS patients harboring familial genetic mutations and control neurons from unaffected individuals. To learn more about their native electrical activity and to simultaneously compare control and ALS neurons from various mutations, the researchers turned to the multiwell Maestro™ microelectrode array (MEA) platform.
iPSC-derived motor neurons are functional
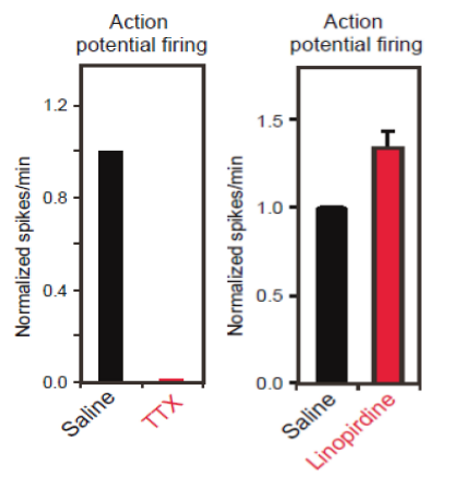
To verify functional activity of ALS iPSC-derived motor neurons, cells were cultured onto Maestro MEA 12-well plates and extracellular voltage was recorded. After 14 days in culture, ALS neurons showed intrinsic electrical activity positively confirming a neural phenotype. When challenged with Tetrodotoxin (TTX), a sodium ion channel blocker, activity was completely eliminated, as expected (raw voltage traces show no neuronal spiking). This reduction in activity was easily quantified using the Maestro software, Axion Integrated Studio (AxIS).
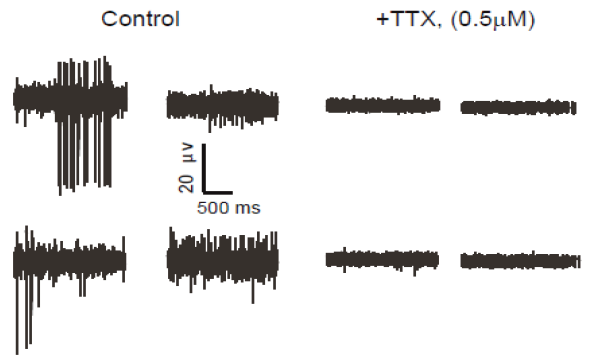
The ALS in vitro model recapitulates clinical results
With the successful generation of functional iPSC ALS neurons, the researchers revealed the enormous possibility that in vitro evaluation of disease models has to offer. To prove the model correlated with clinical findings, the authors used MEA to evaluate iPSC-derived ALS neurons from two patients with the SOD1A4V/+ mutation. It has previously been reported that ALS neurons showed increased neural firing compared to control cells. Maestro recordings confirmed clinical findings of neural hyperexcitability compared to controls.
Below are representative recordings from four out of 64 MEA electrodes in control (11a, 18a) and SOD1A4V/+ mutant (39b, RB9d)-derived motor neurons cultured for 28 days on the arrays. MEA recordings revealed increased spontaneous firing in the mutant-derived neurons compared to control-derived neurons, confirming previous clinical findings and further validating the in vitro model.
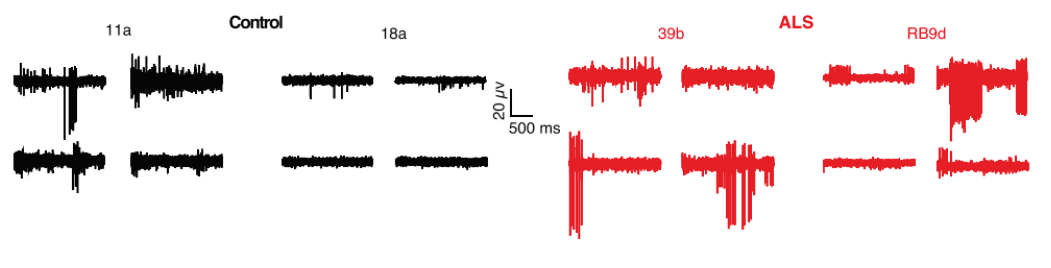
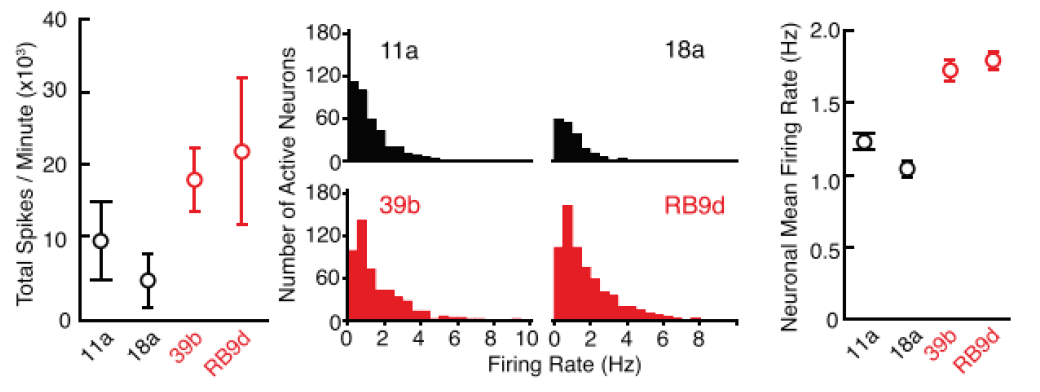
Mean firing rate histograms from individual neurons illustrate phenotypic differences
Spike waveforms from control (below, black) and SOD1A4V/+ (red) neurons were analyzed to derive total action potential firing rate during one minute of recording from MEAs (left), and firing rate histograms of individual neurons (middle). Action potentials were sorted by spike morphology and timing to derive clusters corresponding to individual neurons. The average of mean firing rate histograms of individual neurons from MEAs is illustrated in the right graph, and demonstrates the hyperexcitability of ALS neurons compared to control.
Clinically-relevant iPSCs provide a means to characterize additional ALS mutations
The confirmation of hyperexcitability in the SOD1A4V/+ mutant iPSC neurons validated the in vitro model and led to additional questions about the activity of other ALS-derived neurons. Like many diseases, ALS has numerous disease-causing mutations including SOD1A4V/+, C9orf72, and Fused-in Sarcoma (FUS). Lack of mouse models for the latter two mutations has prevented researchers from examining whether hyperexcitability was specific to SOD1 mutations or a wider trait among many ALS patients. To address this question, the researchers created iPSC neurons from patients harboring each of the mutations. MEA evaluation of each cell type indicated that all ALS iPSC cell lines, independent of mutation, displayed hyperexcitability. The image below illustrates that a variety of SOD1A4V/+, C9orf72, and FUS-derived motor neurons were all hyperexcitable compared to six iPSC lines made from individual healthy controls.
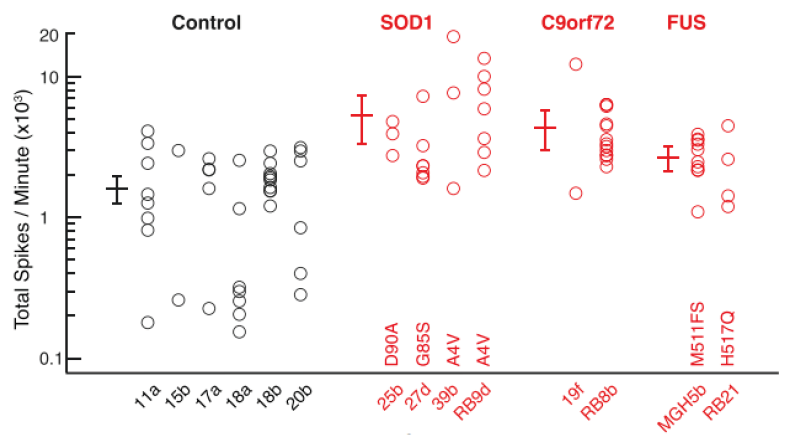
ALS model using iPSC neurons leads to the discovery of a new therapeutic now in clinical trials
After MEA confirmed hyperexcitability across all tested ALS-derived neurons, the researchers began to investigate ways to reverse this phenotype. Earlier data collected in this study showed that a potassium channel blocker could induce hyperexcitability. Could a potassium channel opener do the reverse? Again using MEA as a measure of neural activity, the researchers tested potassium channel openers such as Flupirtine and Retigabine (a clinically-approved anticonvulsant) on iPSC-derived neurons from each mutation. True to their hypothesis, both drugs reduced neuron firing frequency.
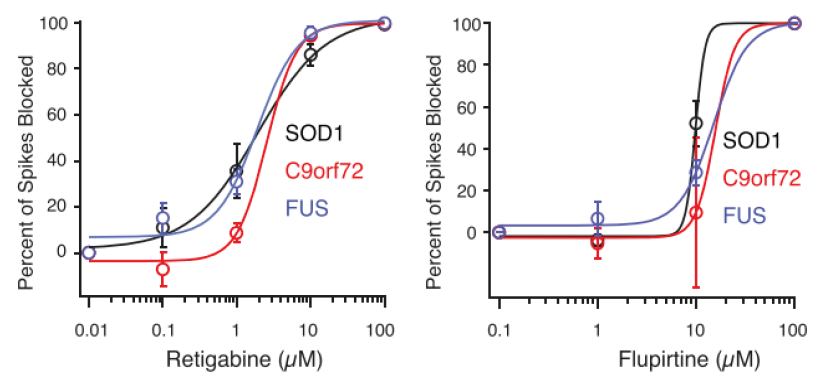
Benefits of using Maestro MEA to characterize and evaluate iPSC-derived motor neurons from healthy controls and ALS patients
- Multiwell Maestro MEA allowed simultaneous recordings from control and ALS-derived neurons for experimental consistency and accuracy.
- Functional information from microelectrode array is captured without dyes or compromising cell integrity providing access to long-term culture and network maturation information in a clinically-relevant in vitro model.
- MEA together with iPSC technology provided insight into cellular function that had previously been unexplored with animal models.
- The ease of evaluating multiple ALS mutation-derived cell lines with MEA allowed researchers to conclude that motor neuron hyperexcitability in ALS may be a distinguishing activity phenotype.
- The ability to reverse this phenotype with approved drugs that calm excitability in neurons led researchers to discover a potential therapeutic for ALS with the help of the Maestro MEA platform.
References
- Wainger et al., “Intrinsic Membrane Hyperexcitability of Amyotrophic Lateral Sclerosis Patient-Derived Motor Neurons,” Cell Reports 7, 1-11 (2014).
- Kiskinis et al., “Pathways Disrupted in Human ALS Motor Neurons Identified through Genetic Correction of Mutant SOD1,” Cell Stem Cell 14, 781-795 (2014).



